Dans le système SMILES de représentation de structures chimiques, l’expression d’un radical est assez simple en apparence, il suffit d’encadrer le centre radicalaire dans des crochets, sauf que …
Publication initiale sur buildblog.buidez.net (2014) – Article mis à jour en Janvier 2025.
Navigation dans le guide
Guide [ Formats moléculaires (chimie) ]
1. Un premier exemple
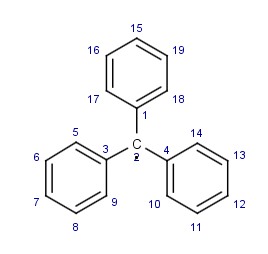
Prenons l’exemple du radical [triphénylméthyle]. ou il sera facile de repérer le centre radicalaire porté par le groupe méthyl dans la chaine de caractères car il est environné par des cycles benzéniques:
 |
 |
|
C1=CC=C(C=C1)[C](C1=CC=CC=C1)C1=CC=CC=C1 |
c1ccc(cc1)[C](c1ccccc1)c1ccccc1 |
Selon que l’aromaticité de la molécule est explicite (figure de droite) ou non (gauche), nous obtenons des chaines SMILES plus ou moins compliquées, mais ou le radical reste exprimé de la même manière. La seconde chaine (droite) correspond à une forme canonique. Deux cycles sont marqués par des numéros 1 (par exemple C1 à C1) qui définissent la zone de fermeture du cycle, les parenthèses sont utilisées pour délimiter un substituant. La manière dont les chaînes SMILES sont générées est surprenante au premier abord, en particulier pour le premier des 3 cycles qui semble être décomposé en deux. Ceci s’explique par le fait que cycle est connecté au C. radicalaire par un atome de carbone qui ne correspond pas à ceux qui sont utilisés (les C1) pour définir la zone de fermeture du cycle.
2. Des extensions
Il existe des tentatives d’extension à la convention SMILES, par exemple le fait d’utiliser des caractères minuscules (Open SMILES) au lieu de majuscules pour spécifier les radicaux. Dans ce cas nous aurions la chaîne Cc pour un radical éthyl. Si nous observons les chaines SMILES produites à partir du triphénylméthyle, nous remarquerons que l’une d’entre elles utilise également des caractères minuscules … pour les atomes impliqués dans des cycles aromatiques. Donc risque de confusion et la nécessité de plus d’intelligence dans le parseur SMILES, sans pour autant garantir un résultat non ambigu.
Il existe d’autres extensions du format SMILES, nous en citerons 2 pour exemple. La convention Daylight et l’extension CXMILES d’origine ChemAxon. La table suivante montre des chaines SMILES générées dans le cas d’une autre molécule:
 |
SMILES | [CH]1C=CCC2=C1C=CC=C2 |
| Canonique | C1=CCc2c([CH]1)cccc2 |
|
| Daylight | [CH]1C=CCc2ccccc12 |
|
| CXSMILES | [CH]1C=CCC2=C1C=CC=C2 |c:1,4,7,9,^1:0| |
Nous constatons que le radical (chaine [CH]) sur l’atome de carbone numéro 1 est bien prise en compte dans les formulations standard et Daylight ainsi que dans la partie classique (à gauche du caractère |) de la formulation CXSMILES. La notation prends cette fois, en tant que centre radicalaire, l’ensemble [CH] et non l’atome seul [C] comme dans le cas précédent.
La forme canonique est calculée avec le programme décrit dans l’article [Décomposition de chaines SMILES] sur ce blog, et à partir de la déclinaison marquée ‘SMILES’. La bibliothèque OpenBabel/Pybel est utilisée dans ce cas. D’ailleurs, la documentation d’OpenBabel présente une page tout à fait intéressante sur la gestion des radicaux avec cette bibliothèque.
Si nous utilisons une formule ou l’aromaticité est explicite, et pour la même molécule, avec la même numérotation des atomes, nous obtenons:
 |
SMILES | [CH]1C=CCc2ccccc12 |
| Canonique | C1=CCc2c([CH]1)cccc2 |
|
| Daylight | [CH]1C=CCc2ccccc12 |
|
| CXSMILES | [CH]1C=CCc2ccccc12 |c:1,^1:0| |
Nous constatons à nouveau une simplification des formulations, la forme canonique restant inchangée. La partie droite de la formulation CXSMILES correspond à une annotation composite (termes séparés par une virgule) qui embarque des métadonnées. Dans ce cas précis, le bloc de métadonnées décrit de manière explicite les doubles liaisons (caractères ^c:) et le centre radicalaire (caractères ^1:). On constate par rapport à la molécule précédente que la partie ^c a également été simplifiée, en effet il ne reste plus qu’une double liaison (C2=C3) à décrire dans la molécule ou l’aromaticité est explicite. Nous reviendrons plus loin sur l’analyse de cette annotation du format CXSMILES.
Nous pouvons aussi nous poser la question d’une formule ou des atomes d’hydrogène sont exprimés de manière explicite. Toujours avec la même molécule, dotée d’une numérotation identique, nous obtenons:
 |
SMILES | [H]C1([H])C=C[CH]C2=C1C=CC=C2 |
| Canonique | C1=CCc2c([CH]1)cccc2 |
|
| Daylight | [CH]1C=CCc2ccccc12 |
|
| CXSMILES | [H]C1([H])C=C[CH]C2=C1C=CC=C2 |c:3,6,9,11,^1:5| |
Nous constatons également qu’il n’y a pas d’ambiguïté car les hydrogènes 11 et 12 sont pris en compte par une chaine [H] qui ne peut pas être confondue avec le [CH]. radicalaire. La forme Daylight évacue le problème des formes hydrogénées ou non et utilise systématiquement les caractères minuscules pour les systèmes aromatiques, délocalisés et doubles liaisons (à titre d’information, une triple liaison s’exprime par le caractère #, C#N pour HCN par exemple). La forme canonique ne prends pas en compte les hydrogènes explicites et reste identique aux deux formulations précédentes.
Par contre, nous constatons que avec une même numérotation de départ, des choses changent dans le bloc de métadonnées CSSMILES. Les numéros, qui correspondent à des numéros d’atomes, ont changé d’une formule développée à une autre: |c:1,4,7,9,^1:0| pour la première formule et |c:3,6,9,11,^1:5| pour la troisième. Pour essayer de comprendre nous devons d’abord aborder la documentation de l’extension ChemAxon CXSMILES (les liens vers les sources d’information originelles sont donnés en fin d’article).
3. Blocs feature de l’extension CXSMILES
L’extension CXSMILES ajoute donc un bloc de métadonnées à la fin de la chaine SMILES, bloc délimité par deux caractères | (pipe). Chaque sous-bloc (feature) à l’intérieur de cet espace est utilisé comme une annotation et est séparé par des virgules. Les radicaux monovalents sont exprimés par ^1, soit la chaîne ^1:0 pour la première molécule et la chaîne ^1:5 pour la seconde. Si nous consultons la documentation CXSMILES relative à l’expression des radicaux nous avons un peu plus de précisions sur ce type d’annotation (citation):
Radical numbers
characters separated by commas. |
Le chiffre qui suit, après le type de radical, est donc sensé correspondre à un numéro d’atome: celui du centre radicalaire, soit l’atome numéro 7. Nous constatons dans les exemples précédents qu’il n’y a pas de correspondance. Avec cette interprétation, nous avons donc plusieurs problèmes concernant l’index du centre radicalaire. D’une part: un numéro zéro, d’autre part la numérotation des deux formes étant identique (à l’exception des deux atomes d’hydrogènes dessinés sur la seconde molécule) nous n’avons pas le même numéro d’atome (0 et 5).
Si nous affichons la chaine MDL Mol (certains atomes sont masqués: ‘…’ pour simplifier) correspondant au premier composé (sans hydrogènes explicites), nous constatons que ce format détermine clairement les charges et les radicaux, à la fin du fichier (lignes commençant par les mots clefs M CHG et M RAD). Dans notre cas, le radical monovalent porté par l’atome de carbone numéroté en septième position est étiqueté ‘7’ et ‘2’, le chiffre ‘2’ signifiant qu’il s’agit d’un radical ou il manque un hydrogène (notation ‘1’ ou ‘3’ pour les carbènes et nitrènes ou il manquera 2 hydrogènes).
|
1 2 3 4 5 6 7 8 9 10 11 12 |
Marvin 12021317572D 10 11 0 0 0 0 999 V2000 -3.2411 0.6188 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -3.9555 0.2063 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 -3.9555 -0.6188 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0 ... 7 10 1 0 0 0 0 8 9 1 0 0 0 0 9 10 2 0 0 0 0 M RAD 1 7 2 M END |
La formule développée est donc bien cohérente, entre la chaine MDL Mol et la numérotation affichée dans l’éditeur. Dans la même voie, nous pouvons nous intéresser à une autre feature dans le bloc de métadonnées. La notation c: indique un numéro d’atome caractérisant une insaturation en CIS. Nous pouvons à nouveau citer la documentation CXSMILES:
 |
Nous constatons que les chaines c:1,4,7,9 et c:3,6,9,11 n’ont pas une partie commune (la numérotation est pourtant identique entre les deux molécules). D’autre part il est difficile d’attribuer (atome a1) une chaine a1-a2=a3-a4 pour chacun des numéros des blocs ‘^c:‘.
4. Une reformulation moléculaire
Par contre, nous pouvons nous demander si en important à nouveau ces chaines CXSMILES dans l’éditeur (fonction ‘copy as’) nous pouvons retrouver une notation cohérente. Dans la table suivante, la première chaine CXSMILES indique la formulation utilisée en import, et celle qui est déduite (fonction ‘copy as’) de la molécule de droite. La dernière partie de la formulation MDL Mol est également indiquée en gras dans la ligne qui suit les chaines SMILES.
|
 |
|||
|
 |
Nous constatons que les importations donnent les mêmes molécules que dans le cas précédent, ce qui montre que la formulation Chemaxon est cohérente (la partie purement SMILES l’est de toutes manières) même si le bloc métadonnées est difficile à comprendre.
Coté description des liaisons en cis, il est encore difficile de réaliser les attributions sur les bases telles qu’elles sont définies dans la documentation. Coté radicaux, nous trouvons une numérotation en n-1 (atome zéro au lieu de 1, et atome 5 au lieu d’atome 6 dans la version partiellement hydrogénée).
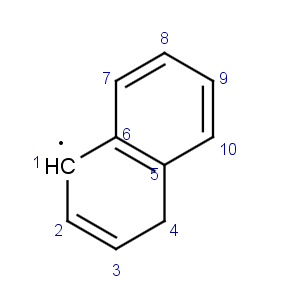
Nous pouvons encore améliorer la cohérence de la structure. Reprenons la molécule précédente en version standard et exprimons d’une autre manière le cycle benzénique: en effectuant simplement un décalage des doubles liaisons, d’une liaison carbone-carbone:
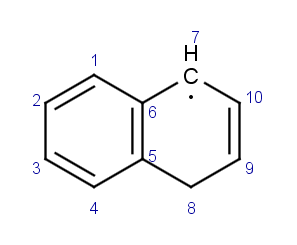 |
devient |  |
|
Nous avons le même problème de numérotation concernant le centre radicalaire que dans l’exemple précédent. Nous allons utiliser le même protocole pour générer une molécule dont la numérotation est cohérente. La table suivante montre la nouvelle molécule, on constate que nous n’avons pas encore corrigé la numérotation du centre radicalaire mais que nous progressions sur le cas des liaisons en cis et trans:
 |
 |
|
Les parties colorées en vert servent à identifier des assignations a1-a2=a3-a4 en cis ou trans. Prenons le cas des doubles liaisons en cis, nous avons c:1,6 dans le bloc de métadonnées, ce qui indique que l’on doit avoir c1-c2=c3-c4 et c6-c7=c8-c9 ce qui est bien le cas.
En ce qui concerne les liaisons en trans, nous retrouvons également une numérotation cohérente, t:4,8 donne c4-c5=c6-c7 et c8-c9=c10-x ou x correspond à c1.
D’autre part, nous constatons que la dernière numérotation ne correspond pas tout à fait à la règle a1-a2=a3-a4 ou chaque atome correspond à un numéro d’atome croissant, puisque x=c1. Dans ce cas on dirait qu’il n’y a pas vraiment de solution, mais il reste quand même une option, celle de simplifier la formulation en utilisant une notation différente pour le cycle benzène:
 |
|
Nous sommes directement passés à la formule dont la numérotation est cohérente (export puis (re)import de la chaine CXSMILES). Nous constatons que la liaison en cis qui se numérote à partir de l’atome de carbone numéro 1 répond au schéma c1-c2=c3-c4 de manière correcte (ou en tout cas telle que définie dans la documentation). La simplification induite par l’utilisation de l’aromaticité porte donc ses fruits.
Si nous utilisons la forme canonique de cette molécule ‘C1=CCc2c([CH]1)cccc2‘ nous constatons une intégration sans problèmes, et surtout une conservation de la numérotation en import/export. Cette numérotation est identique au cas précédent (démarrage par le C. en 1, et atomes 5; 10 à la jonction entre cycles). Il est vrai que la forme canonique aromatise systématiquement le cycle benzénique.
5. L’atome zéro
Nous allons examiner le problème de l’atome zéro utilisé pour l’annotation du centre radicalaire en utilisant une autre technique. Nous allons simplement exprimer l’hydrogène porté par le carbone numéroté en 1, au moyen d’une liaison (covalente) formelle, plutôt que de laisser à l’éditeur le soin de l’exprimer de manière implicite. Ce qui nous donnera:
 |
|
Nous constatons qu’il n’y a pas d’impact négatif sur l’expression de la double liaison en cis, mais que la chaine ^1:0 s’est transformée en chaîne ^1:1. L’index d’atome qui suit le centre radicalaire indiquerait donc un atome consécutif et relié au centre, dans le cas ou l’hydrogène est implicite, le numéro zéro est utilisé, dans le cas ou il est explicite le numéro de l’atome H est exprimé. Si nous remplaçons cet hydrogène H1 par un groupe méthyl nous obtenons la chaine CSXMILES suivante C[C]1C=CCc2ccccc12 |c:2,^1:1| ce qui semble confirmer l’idée.
Pourtant, nous n’en avons pas encore fini … si nous examinons à nouveau la première molécule de l’article (le radical triphénylméthyle), une fois le processus d’exportation/(re)importation réalisé, nous obtenons:
 |
c1ccc(cc1)[C](c1ccccc1)c1ccccc1 |^1:6| |
L’atome numéro 6 est situé sur un cycle benzénique et pas vraiment sur le carbone central qui est numéroté en 7. Donc la notation ^1:6 réfèrerait plutôt à une désignation en n-1, soit ^1:(n-1) ou n correspond à l’index du centre radicalaire. En d’autres termes (informatiques) le numéro qui suit la notation ^1: est à comprendre comme un indice [0 .. n-1] et non comme un index [1 .. n].
Nous pourrons le vérifier avec des dérivés plus compliqués, tels que [DMPO-OH]. ou l’oxygène en 9 est porteur du radical (chaine CXSMILES CC1(C)CCC(O)N1[O] |^1:8|) ou [TPNO]. ou l’oxygène en 12 est porteur du radical (chaine CC1(C)CC(O)CC(C)(C)N1[O] |^1:11|) qui sont utilisés dans différentes applications (spin trapping, antioxydants).
6. Conclusion
Cet article donne l’impression de couper les cheveux en 4, mais il rends compte de subtilités qui passent généralement au dessus de la plupart des utilisateurs de ce format. Subtilités dont la prise en compte peut être intéressante dans le début d’un processus de conception, pour ne pas aller chercher le bug impossible plus tard lors de l’exploitation du code ou de l’application.
La recherche des formes canoniques devrait être intégrée dans la gestion des bibliothèques de molécules, car elle amène des avantages en termes de gestion du format chimique, même si la perte de doubles liaisons explicites, en particulier des hétérocycles, peut gêner l’œil du chimiste. Coté modélisation (au moins pour les processus de drug design) la plupart des molécules qui entrent dans des ligandothèques sont très souvent systématiquement aromatisées pour des raisons d’interprétation par les programmes de calcul.
Liens et lectures
- SMILES [ http://fr.wikipedia.org/wiki/Simplified_Molecular_Input_Line_Entry_Specification ].
- Daylight: SMILES – A Simplified Chemical Language [ http://www.daylight.com/dayhtml/doc/theory/theory.smiles.html ].
- Open SMILES [ http://opensmiles.org/ ].
- ChemAxon Extended SMILES and SMARTS (CXSMILES and CXSMARTS) [ https://docs.chemaxon.com/display/docs/formats_chemaxon-extended-smiles-and-smarts-cxsmiles-and-cxsmarts.md ].
- OpenBabel: Radical and SMILES extensions [ https://openbabel.org/docs/Features/Radicals.html ].
- A Drefahl. CurlySMILES: a chemical language to customize and annotate encodings of molecular and nanodevice structures. Journal of Cheminformatics (2011) 3:1, 2-7. DOI [ 10.1186/1758-2946-3-1 ].
- Je remercie G. Dupuis pour son site remarquable et les exemples fournis, que j’ai utilisés.
- G. Dupuis – Cours de chimie générale et organique [ https://chimieorganique.faidherbe.org/ ].