Le module pdb.distance fournit des fonctions de base qui analysent les distances entre atomes, groupes d’atomes et résidus. Ces fonctions peuvent servir de primitives pour construire des requêtes 3D plus évoluées, et qui correspondront à quelques questions que pourraient se poser des modélisateurs ou des biologistes structuraux. Cet article traite les fonctions de base du module distance.py et de leur intégration avec les structures de données (pdbm, resmap) du paquetage, c’est ce qui en fait leur puissance. Un autre article fera le point sur l’intersection de requêtes 3D en utilisant cette méthodologie, un troisième article donnera des éléments concernant une structure de données spécialisée : la proxmap.
Publication initiale sur buidez.net (2014) – Article mis à jour en Avril 2025.
Navigation dans le guide
Guide [ Composants : buildez.pdb ]
1. Estimateurs de distance : 2 atomes
La primitive de base consiste à mesurer la distance entre deux atomes. La fonction pdb_atom_dist implémente un calcul simple de distance euclidienne en 3 dimensions, mais pas n’importe comment. Au lieu d’accepter comme argument des valeurs x, y, z de coordonnées ou des vecteurs (x,y,z)r et (x,y,z)q pour deux atomes (reference, query), cette fonction prends en charge deux lignes atomiques d’une matrice pdbm. Il est également possible de fournir directement deux lignes atomiques d’un flot PDB sous la forme de deux chaines de caractères puis de les transformer individuellement en lignes de matrices pdbm avec la fonction pdb_coordlinetyped_get (ou la fonction pdb_str2pdbl de pdb.pdbm) que l’on peut trouver dans le module pdb.str et qui est utilisé dans la construction interne des matrices pdbm (typiquement les fonctions pdb_str2pdbm ou pdb_strvec2pdbm).
Une fois que l’on sait calculer la distance entre deux atomes, est ce qu’ils sont ‘près’ ou ‘loin’ ?
Et c’est cette question qui est importante, et qui a du sens (biochimique). Cet aspect est pris en charge par les fonctions pdb_atom_isnear et pdb_str_atom_isnear qui utilisent respectivement des lignes atomiques de matrices pdbm (query, reference) ou de chaines de caractères en tant qu’arguments. La valeur (score) retournée sera 0 (considérée comme ‘loin’) ou 1 (comme ‘près’) en fonction d’une minimum passée comme argument (5 Angströms par défaut).
Les fonctions pdb_atom_isnearcub et pdb_atom_isnearcub2 réalisent la même opération que pdb_str_atom_isnear et pdb_atom_isnear et mais en utilisant une approximation cubique (et non un calcul de distance) dans l’espace de coordonnées cartésiennes, ce qui donne parfois une bonne approximation et permet d’accélérer les calculs.
Application: structure 1HXD
On va prendre comme exemple des éléments de la structure 1HXD [Weaver_2001]. Cette structure présente deux chaines polypeptidiques (A, B). Chaque chaine est associée avec une molécule de Biotine (nom de résidu BTN).
L’exemple montre simplement comment évaluer la proximité entre deux atomes (a1, a2) d’une même Biotine (résidu 500) ou entre un atome de la biotine 500 (a1) et un autre (a3) de la Biotine 501 qui est dans l’autre chaîne polypeptidique.
Dans le premier cas les atomes doivent être proches, dans l’autre ce n’est probablement pas le cas.
Le code suivant permet d’avoir une idée de l’utilisation de ces primitives:
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 |
print("************************************************************************") print("*** Test two atoms distances") print("************************************************************************") print("given two atoms of the same ligand") a1 = "HETATM 4715 C11 BTN 500 55.016 4.107 53.277 1.00 24.55 C " a2 = "HETATM 4716 O11 BTN 500 53.849 4.252 52.886 1.00 21.58 O " print("a1 [%s]" % (a1)) print("a2 [%s]" % (a2)) print("and one from another subunit") a3 = "HETATM 4731 C11 BTN 501 3.898 2.712 54.208 1.00 25.27 C " print("a3 [%s]" % (a3)) print() print("=> test pdb_atom_isnearcub at 5.0 Angstrom") print("a2 in a1 ? Result is %d" % (pdb_atom_isnearcub(a1, a2, 5.0))) print("a3 in a1 ? Result is %d" % (pdb_atom_isnearcub(a1, a3, 5.0))) print() print("=> test pdb_atom_isnear at 5.0 Angstrom") la1 = pdb_coordlinetyped_get(a1) la2 = pdb_coordlinetyped_get(a2) la3 = pdb_coordlinetyped_get(a3) print("a2 in a1 ? Result is %d" % (pdb_atom_isnear(la1, la2, 5.0))) print("a3 in a1 ? Result is %d" % (pdb_atom_isnear(la1, la3, 5.0))) print() print("=> test real distances (in Angstrom)") print("d(a2,a1) = %f" % (pdb_atom_dist(la1, la2))) print("d(a3,a1) = %f" % (pdb_atom_dist(la1, la3))) |
La figure suivante montre la structure 1HXD avec ses deux chaînes et les distances entre les atomes C11 et O11 de BTN500 (première chaîne à droite, 1.239 Å) et entres les atomes C11 de BTN500 (première chaîne, à droite) et C11 de BTN501 (seconde chaîne à gauche, BTN501 colorée en jaune, 51.146 Å).
 |
Le résultat à l’exécution sera le suivant :
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 |
************************************************************************ *** Test two atoms distances ************************************************************************ given two atoms of the same ligand a1 [HETATM 4715 C11 BTN 500 55.016 4.107 53.277 1.00 24.55 C ] a2 [HETATM 4716 O11 BTN 500 53.849 4.252 52.886 1.00 21.58 O ] and one from another subunit a3 [HETATM 4731 C11 BTN 501 3.898 2.712 54.208 1.00 25.27 C ] => test pdb_atom_isnearcub at 5.0 Angstrom a2 in a1 ? Result is 1 a3 in a1 ? Result is 0 => test pdb_atom_isnear at 5.0 Angstrom a2 in a1 ? Result is 1 a3 in a1 ? Result is 0 => test real distances (in Angstrom) d(a2,a1) = 1.239272 d(a3,a1) = 51.145505 |
On en conclue donc que a1 (BTN.500.C11) est ‘proche’ (distance inférieure ou égale à 5 Å) de a2 (BTN.500.O11) et que a1 est ‘éloigné’ de a3 (BTN.501.C11).
Pour des distances élevées l’approximation cubique est évidemment efficace, il y a près de 50 Å de distance entre chaque site de liaison Biotine-Enzyme.
2. Estimateurs de distances : blocs d’atomes
Par groupe d’atomes on peut imaginer un ‘paquet’ d’atomes plus ou moins typé, mais qui peut aller jusqu’à l’enveloppe d’un site actif, un élément structural (hélice, boucle …), un ligand, un résidu ou une combinaison de ces ensembles (par exemple un site actif qui serait défini par une liste de résidus).
Maintenant que l’on sait comment faire pour savoir si deux atomes sont ‘proches’ ou non, on peut se poser la même question pour des groupes d’atomes.
La première fonction capable de réaliser ce type de calculs est pdb_pdbm_block_isnear qui accepte en entrée deux matrices pdbm et la limite en distance (toujours 5 Å par défaut). Son utilisation est très simple, le code suivant montre comment charger le fichier 1HXD dans une chaine de caractères s, puis en extraire le ligand (BTN500) et la chaine A (chaines de caractères s1 et s2), et transformer ces deux blocs en deux matrices pdbm (m1, m2).
Préparation des données
Les requêtes d’extraction opérant en mode string, sur des matrices pdbm, des resmap qui sont implémentées dans le paquetage buildez.pdb permettent de fournir les ensembles de coordonnées (query, reference) sans trop de problèmes.
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 |
print("************************************************************************") print("*** Preparing data for tests with groups of atoms") print("************************************************************************") print("loading coordinates (only) of [1hxd.pdb] structure") s = pdb_file2str(file_cleanpath(site_package + '/test/1hxd.pdb')) s = pdb_str_key_get(s,"ATOM HETATM") m0 = pdb_str2pdbm(s) print("get some parts of structure as strings (using str_pdb and str_filter modules)") print("=> get the protein (s1) corresponding to 'A' chain") s1 = pdb_str_coordsblock_get(s, "ATOM", pdb_field_coords_id("CHAINID"), "A", "=", 0) m1 = pdb_str2pdbm(s1) print("=> get the ligand (s2) corresponding to Ligand 500 (Biotin, 'A' chain)") s2 = pdb_str_coordsblock_get(s,"HETATM",pdb_field_coords_id("RESSEQ"),"500","=", 0) m2 = pdb_str2pdbm(s2) print("=> get the ligand (s3) corresponding to Ligand 501 (Biotin, 'B' chain)") s3 = pdb_str_coordsblock_get(s,"HETATM",pdb_field_coords_id("RESSEQ"),"501","=", 0) m3 = pdb_str2pdbm(s3) print("=> get all the water (s4) we can do") s4 = pdb_str_coordsblock_get(s,"HETATM",pdb_field_coords_id("RESNAME"),"HOH","=", 0) |
Rappelons que la fonction pdb_pdbm2resmap du module pdb.residue transforme une matrice pdbm en resmap, et qu’il existe également de quoi transformer une chaine de caractères PDB en matrice pdbm dans le paquetage buildez.pdb.
Calcul de distances
Puis la fonction pdb_pdbm_block_isnear est appelée (à 3 Å de distance) et renvoie deux valeurs (cn, cq). Le code correspondant pourrait être :
|
1 2 3 4 5 6 7 8 9 10 11 |
print("************************************************************************") print(" Working with distance estimators for groups of atoms => test 1 ") print("************************************************************************") print("*** checking with pdbm matrixes") print("=> ref is BTN500, query is chain A, 3 angstroms") print("Length of query before operation is %d atoms" % (len(m1))) (cn, cq) = pdb_pdbm_block_isnear(m1, m2, 3.0, 1) print("Number of atoms found is %d" % (cn)) if (cn > 0): for i in range (0, len(cq), 1): print(cq[i]) |
Ce qui va nous donner (les deux codes confondus):
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 |
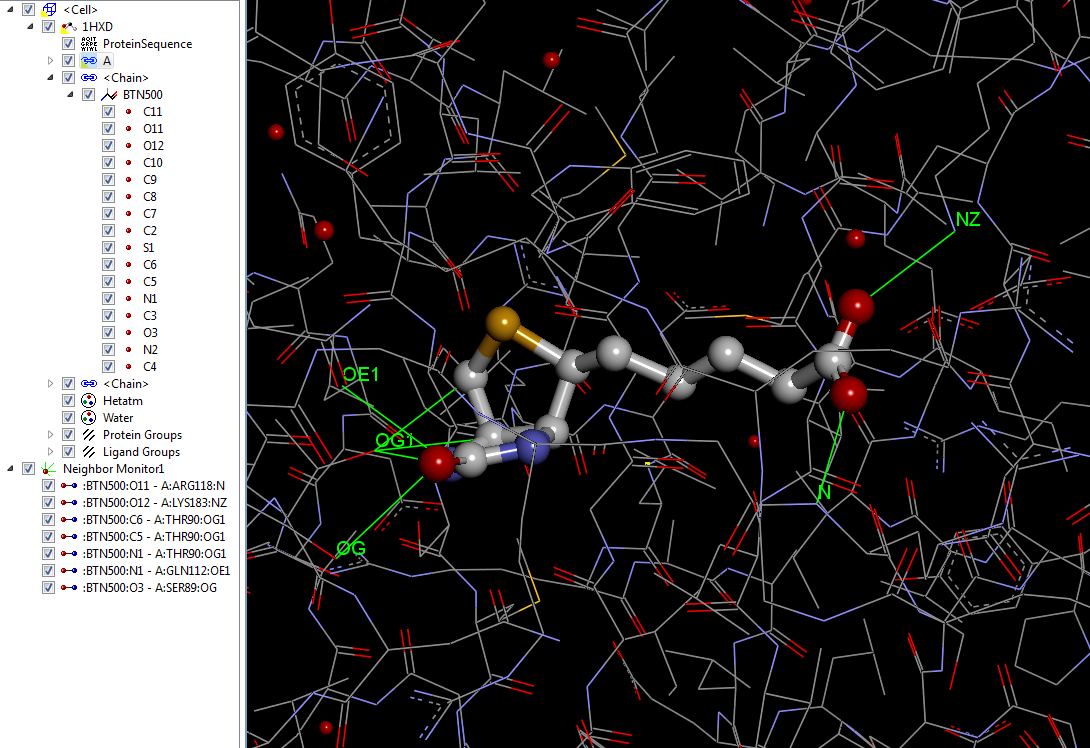
************************************************************************ *** Preparing data for tests with groups of atoms ************************************************************************ loading coordinates (only) of [1hxd.pdb] structure get some parts of structure as strings (using str_pdb and str_filter modules) => get the protein (s1) corresponding to 'A' chain => get the ligand (s2) corresponding to Ligand 500 (Biotin, 'A' chain) => get the ligand (s3) corresponding to Ligand 501 (Biotin, 'B' chain) => get all the water (s4) we can do ************************************************************************ Working with distance estimators for groups of atoms => test 1 ************************************************************************ *** checking with pdbm matrixes => ref is BTN500, query is chain A, 3 angstroms Length of query before operation is 2356 atoms Number of atoms found is 5 ['ATOM', 643, 'OG', '', 'SER', 'A', 89, '', 58.058, 13.097, 47.232, 1.0, 21.6, '', 'O', '', 643] ['ATOM', 649, 'OG1', '', 'THR', 'A', 90, '', 62.362, 11.104, 48.329, 1.0, 21.66, '', 'O', '', 649] ['ATOM', 823, 'OE1', '', 'GLN', 'A', 112, '', 60.861, 12.645, 50.225, 1.0, 30.02, '', 'O', '', 823] ['ATOM', 858, 'N', '', 'ARG', 'A', 118, '', 52.239, 5.387, 51.005, 1.0, 27.4, '', 'N', '', 858] ['ATOM', 1394, 'NZ', '', 'LYS', 'A', 183, '', 54.724, 2.06, 56.418, 1.0, 12.21, '', 'N', '', 1394] |
La première valeur cn correspond au nombre d’atomes qui ont satisfait à la requête et la seconde valeur cq correspond à la matrice pdbm qui regroupe ces mêmes atomes.
Nous constatons que 5 atomes correspondent à la requête, correspondant chacun à 5 résidus différents.
La figure suivante affiche le même type de requête avec un logiciel standard du marché (Discovery Studio) et qui allume les mêmes atomes à 3 Å de distance :
 |
On peut étendre la fonction pdb_pdbm_block_isnear de manière à utiliser directement des morceaux de fichiers PDB. Les deux blocs peuvent êtres passés sous forme de chaines de caractères (incluant les caractères de fin de ligne) avec la fonction pdb_str_block_isnear ou directement sous forme de listes Python de chaines de caractères (une ligne de fichier PDB par élément de liste) avec la fonction pdb_vec_block_isnear. Les trois fonctions peuvent être utilisées en mode strict (strict=1 par défaut) ou non (strict=0). Dans le second cas l’approximation cubique est utilisée au lieu du calcul direct de distance.
3. Estimateurs de distances : résidus
Une fois que l’on sait faire pour un groupe d’atomes, il est facile de l’étendre à des résidus, et de répondre à la question :
Quels sont les résidus proches d’un atome ou d’un groupe d’atomes (bloc, résidu, ligand …) ?
Ces opérations sont réalisées par les fonctions pdb_pdbm_blockres_isnear, pdb_vec_blockres_isnear et pdb_str_blockres_isnear.
La première utilise des matrices pdbm pour définir les blocs d’atomes (query, reference), la seconde deux listes de chaînes de caractères, la troisième deux chaînes de caractères (incluant les caractères de fin de ligne). Les trois demandent une distance de test et fonctionnent en mode strict (strict=1 par défaut, calcul direct) ou non (approximations cubiques). Les trois renvoient une resmap, ce qui est logique (et pratique) car on demande un résultat qui est exprimé en termes de résidus et non d’atomes.
Prenons un premier code pour montrer l’utilisation de pdb_pdbm_blockres_isnear :
|
1 2 3 4 5 6 |
print("=> check for a resmap of residues near BTN500 (at 3 A)") r1 = pdb_pdbm_blockres_isnear(m1, m2, 3.0, 1) print("number of residues found %d" % (len(r1))) if (len(r1) > 0): for i in range (0, len(r1), 1): print(r1[i]) |
On utilise les mêmes matrices pdbm (m1, m2) que dans l’exemple précédent, et le résultat sera comparable, mais exprimé sous la forme d’une resmap :
=> check for a resmap of residues near BTN500 (at 3 A)number of residues found 5['SER', 89, 643, 643, -1, '']['THR', 90, 649, 649, -1, '']['GLN', 112, 823, 823, -1, '']['ARG', 118, 858, 858, -1, '']['LYS', 183, 1394, 1394, -1, ''] |
Nous retrouvons donc les mêmes résidus que dans la matrice pdbm (valeur cq) qui était renvoyée par la fonction pdb_pdbm_block_isnear.
En mode texte (string)
Les fonctions en mode texte sont faciles à utiliser, par exemple pour la fonction pdb_str_blockres_isnear en utilisant une consigne à 3 ou 5 Å :
|
1 2 3 4 5 6 7 8 9 10 |
print("*** checking with strings") print("=> what residues of chain 'A' (query) are near ligand (s1 as reference) ?") print("at 3.0 Angstrom :") r1 = pdb_str_blockres_isnear(s2, s1, 3.0, 1) print("s1 (query) near s2 (ref) => found %d residues" % (len(r1))) print(r1) print("at 5.0 Angstrom :") r1 = pdb_str_blockres_isnear(s2, s1, 5.0, 1) print("s1 (query) near s2 (ref) => found %d residues" % (len(r1))) print(r1) |
Ce qui va donner comme résultat:
|
1 2 3 4 5 6 7 8 |
*** checking with strings => what residues of chain 'A' (query) are near ligand (s1 as reference) ? at 3.0 Angstrom : s1 (query) near s2 (ref) => found 5 residues [['SER', 89, 1, 1, -1, ''], ['THR', 90, 2, 2, -1, ''], ['GLN', 112, 3, 3, -1, ''], ['ARG', 118, 4, 4, -1, ''], ['LYS', 183, 5, 5, -1, '']] at 5.0 Angstrom : s1 (query) near s2 (ref) => found 22 residues [['SER', 89, 1, 4, -1, ''], ['THR', 90, 5, 10, -1, ''], ['ASN', 91, 11, 16, -1, ''], ['GLN', 112, 17, 20, -1, ''], ['ALA', 114, 21, 21, -1, ''], ['GLY', 115, 22, 24, -1, ''], ['ARG', 116, 25, 30, -1, ''], ['GLY', 117, 31, 34, -1, ''], ['ARG', 118, 35, 44, -1, ''], ['ARG', 121, 45, 47, -1, ''], ['TRP', 123, 48, 57, -1, ''], ['TYR', 132, 58, 63, -1, ''], ['LEU', 133, 64, 67, -1, ''], ['SER', 134, 68, 69, -1, ''], ['ASP', 176, 70, 71, -1, ''], ['LYS', 183, 72, 74, -1, ''], ['GLY', 186, 75, 78, -1, ''], ['ILE', 187, 79, 82, -1, ''], ['LEU', 188, 83, 87, -1, ''], ['GLY', 204, 88, 91, -1, ''], ['ALA', 205, 92, 95, -1, ''], ['GLY', 206, 96, 97, -1, '']] |
Forcément, en augmentant la consigne, on allume plus de résidus.
4. Référentiel
Fonctions du module distance.py en mode abrégé (avril 2025).
| Fonction | Utilisation |
pdb_atom_dist |
Calcul de la distance Euclidienne entre les vecteurs (x,y,z)r et (x,y,z)q (reference, query) exprimés sous la forme de lignes (rows) d’une pdbm. |
pdb_atom_isnear |
Utilise les mêmes vecteurs que pdb_atom_dist mais renvoie un score (0 ou 1) si on considère que les atomes sont ‘proches’ au sens biochimique. |
pdb_str_atom_isnear |
Identique à pdb_atom_isnear mais utilise deux chaines (string) PDB comme arguments pour les vecteurs. |
pdb_atom_isnearcub2 |
Identique à pdb_atom_isnear mais utilise une approximation cubique. |
pdb_atom_isnearcub |
Identique à pdb_atom_isnearcub2 mais utilise deux chaines (string) PDB comme arguments pour les vecteurs. |
pdb_pdbm_block_isnear |
Calcul d’une distance en utilisant deux blocs d’atomes au format pdbm. Utilise pdb_atom_isnear ou pdb_atom_isnearcub2. Renvoie un couple avec le score (0 ou 1, meilleure proximité possible) et un bloc pdbm (bloc référence) des atomes impliqués dont la distance est considérée comme ‘proche’. |
pdb_vec_block_isnear |
Utilise deux listes (lignes) de chaines (string) d’un fichier PDB pour faire la même chose que pdb_pdbm_block_isnear. Mais ne renvoie que le bloc d’intersection sous la forme d’une liste de chaines (string) PDB. |
pdb_str_block_isnear |
Interface texte (string) à pdb_vec_block_isnear, entrées et sorties sont au format chaines (avec "\n" en tant que séparateurs de lignes PDB) de caractères. |
pdb_pdbm_blockres_isnear |
Utilise pdb_pdbm_block_isnear mais envoie une resmap au lieu d’un couple (score, pdbm) des atomes impliqués. Peut utiliser des distances Euclidiennes ou une approximation cubique. Ce qui veut dire qu’il s’agit aussi d’une fonction de proximité de résidus, basée sur des atomes qui peuvent faire partie de plusieurs résidus, mais sans nécessité d’impliquer l’ensemble des atomes de ces résidus. |
pdb_vec_blockres_isnear |
Similaire à pdb_pdbm_blockres_isnear, mais utilise en entrée des listes de lignes PDB, renvoie une resmap. |
pdb_str_blockres_isnear |
Interface texte (string) à pdb_vec_blockres_isnear, entrées et sorties sont au format chaines (avec "\n" en tant que séparateurs de lignes PDB) de caractères. |
5. Conclusion
Ces outils d’extraction d’atomes ou de groupes d’atomes sur conditions de distances sont très intéressants. Si nous les combinons à des fonctions destinées à analyser la présence de liaisons covalentes ou d’identification de résidus, nous pourrons peut disposer d’un système capable de traiter de manière automatique des structures protéiques sur la base d’ensembles.
Ces structures de données (pdbm, resmap …) dont on pourrait penser qu’elles sont simplistes, redondantes ou inutiles, simplifient en fait grandement la programmation, car elles sont conçues pour être cohérentes entre elles. D’autre part comme les fonctions, à leur tour, génèrent une matrice pdbm ou une resmap, la régénération d’une chaine de caractères PDB (utilisable dans un autre logiciel) est immédiate pour peu que l’on soit capable de remonter à la chaîne de caractères initiale. Ce qui est possible, car les matrices pdbm embarquent le numéro de ligne dans le fichier PDB d’origine qui correspond à chaque ligne de coordonnées moléculaires.
Il faut noter que les fonctions de ce module sont utilisées pour traiter globalement un bloc d’atomes, il n’y a pas de notions de centrage (centroïde par exemple) à propos des groupes d’atomes, ce type de fonction fait partie d’un autre paquetage de buildez.pdb.
Liens et lectures
- Structure 1HXD [ https://www.rcsb.org/structure/1HXD ].
- Vitamine B8 (biotine) [ https://fr.wikipedia.org/wiki/Vitamine_B8 ].
- Biotinylation [ https://en.wikipedia.org/wiki/Biotinylation ].