Le fichier script.mvdscript est important pour beaucoup de raisons, la première est que nous pouvons le modifier et lancer des calculs avec le même paramétrage sans passer par l’interface utilisateur (GUI) de Molegro. Ce fichier peut être généré de différentes manières : à la main, par un outil informatique maison, par l’interface (GUI) utilisateur de Molegro. Une fois généré par le GUI (pour éviter les erreurs de syntaxe) nous pouvons le modifier dans le texte (valeur des paramètres, éditeur de texte ASCII) de manière à ajuster les calculs jusqu’à ce que la modélisation fonctionne (robustesse, répétabilité, cohérence des résultats).
Publication initiale sur buildblog.buidez.net (2021) – Article mis à jour en Mars 2025.
Navigation dans le guide
Guide [ Aide Molegro (docking) ]
1. Le fichier script et les paramétrages
Ce fichier rassemble des couples clés-valeurs et des blocs (ex : DOCKSETTINGS) concernant le paramétrage des calculs et l’espace de travail (Unnamed_complex.mvdml par défaut) à utiliser. Les lignes qui commencent par // (remarques) ne seront pas utilisées dans le calcul :
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 |
// Molegro Script Job. // Created: ven. 22. janv. 19:14:45 2021 // Structure file: Unnamed_complex.mvdml PARSERSETTINGS breakUnrealisticBonds=false;combineMoleculeFragments=true;useSybylForHybridization=true DOCKSETTINGS maxIterations=10000;runs=20;ignoreSimilarPoses=false;IgnoreSimilarPosesThreshold=1;MaxPoses=5;MinimizeReceptor=4000,2000;poseOutput=mvdml;createSmiles=true;postMinimize=true;postOptimizeHBonds=true POSENAMEPATTERN [$ID] $NAME EVALUATORTYPE MolDockGrid EVALUATOR cropdistance=0;gridresolution=0.30;ligandes=true;sp2sp2bond=true;internalhbond=true;hbond90=true;SoftenPotential=true;DisplaceWater=false;tabuclustering=true,2,100,id OPTIMIZERTYPE MSE OPTIMIZER populationsize=50;cavity=true;creationEnergyThreshold=100;poseGenerator=10,10,30;recombine=true;maxsimplex=1500;simplexsteps=300;simplexdistancefactor=1;clusterthreshold=0.0 TEMPLATE strength=-500;useGrid=true;gridResolution=0.4 LOAD "Unnamed_complex.mvdml" RMSD Ligand[0] DOCK Ligand[0] NEW |
Une partie du paramétrage du calcul est située dans DOCKSETTINGS, notamment le nombre d’itérations maximum (maxIterations) par run (10000) et le nombre de runs (20). Dans ce type de calcul (d’autres paramétrages sont possibles) le nombre de runs correspond au nombre de poses retenues après calcul. Les poses correspondent à un échantillonnage (sur des milliers de calculs) des conformations possibles du ligand associées à un score final (ex: MolDock), différent de celui qui est utilisé pendant la phase de recherche (ex: MolDockGrid). A première vue, il s’agit de la même fonction de score: MolDock. Sauf que celle qui est utilisée pendant la phase de recherche est approximée: elle est discrétisée sur une grille (gridresolution=0.30, exprimée en angströms) de manière à accélérer les calculs.
Nous lisons que l’algorithme de recherche sera basé sur l’optimiseur MSE (OPTIMIZERTYPE), associé à un paramétrage défini dans la section (OPTIMIZER). La fonction de score (EVALUATORTYPE) utilisée pendant la phase de recherche sera MolDockGrid, dont le paramétrage est en partie groupé dans le bloc EVALUATOR. Dans ce bloc nous trouvons également un paramétrage pour le clustering tabuclustering=true,2,100,id qui à pour objet la différentiation des poses. Pour faire simple, sur ce paramétrage, la valeur 2.0 est exprimée en angströms, plus nous la baissons, plus les meilleures poses auront tendance à être identiques.
A la suite, nous trouvons les paramétrages pour des templates (pharmacophores), s’ils sont activés (section TEMPLATE) et d’autres contraintes (non utilisées ici). A noter que l’essentiel du paramétrage des pharmacophores est stocké dans le workspace, nous n’avons ici que 3 paramètres liés au calcul.
Enfin, la ligne DOCK indique quel est le ligand qui va être calculé, ici il s’agit du ligand dont l’indice est zéro dans le workspace Unnamed_complex.mvdml.
Et la protéine ?
En fait elle est incluse dans le workspace Unnamed_complex.mvdml comme les ligands et les autres paramétrages (flexibilité, espace de recherche, templates/pharmacophores, contraintes …) et n’apparaît pas dans le fichier script. L’avantage c’est qu’on peut utiliser ces fichiers .mvdml d’une protéine à l’autre, sans faire beaucoup de modifications (elles s’appellent toutes par défaut Unnamed_complex, sauf si nous décidons autre chose). Une fois que nous avons compris l’essentiel, ce fichier est très pratique à modifier, par exemple augmenter le nombre d’itérations si nous nous apercevons que les calculs sont arrêtés avant la convergence.
Ajouter des ligands
Le script est encore plus pratique si nous pouvons l’utiliser sans rien changer (paramètres identiques et même workspace) pour tous les ligands. Il suffira de copier les fichiers Unnamed_complex.mvdml et script.mvdscript dans un nouveau répertoire, changer l’indice du ligand dans la ligne DOCK et de lancer le calcul. Plus précisément, si nous ajoutons des ligands, il faudra: i) les intégrer dans frame_06_1P45aNAD_Flex19_tmpl.mvdml à la racine, puis ii) sauver cet espace de travail avec un numéro de version, par exemple frame_06_1P45aNAD_Flex19_tmpl-01.mvdml et enfin iii) de copier ce fichier sous le nom Unnamed_complex.mvdml dans le nouveau répertoire. Si l’indice du ligand est 37 dans Unnamed_complex, il suffira de modifier la ligne DOCK Ligand[0] en DOCK Ligand[37] puis de lancer les calculs.
La ligne RMSD
Le JPL est un ligand co-cristallographié dans la structure 3FNG [Freundlich_2009]. Nous nous en servons sert comme d’un étalon interne dans les calculs, de manière à voir si on peut reproduire sa conformation cristallographique originale (3FNG_) dans 1P45a, [Kuo_2003] sachant que les deux sites sont assez similaires (ils font partie d’un même cluster structural dans notre classification gen3) et que la flexibilité des résidus est activée. On calcule donc les RMSD (Root Mean Square Deviation) entre la conformation cristallographique et le ligand par la ligne RMSD Ligand[0].
Ligne que l’on supprimera dans le cas d’un nouveau ligand, car nous n’avons plus de référence à comparer.
2. Lancement de calculs par script
Les calculs peuvent être lancés via le GUI, mais une fois que le paramétrage est stable, nous avons tendance à lancer les calculs via le script. Selon l’OS (c’est possible sous MS Windows) le double clic sur le fichier (ou un appel en mode console) permet de lancer le calcul, dans ce cas une fenêtre à plusieurs onglets apparaît :
 |
Les lignes de log défilent, les autres onglets (images suivantes) permettent de voir les résultats du calcul, soit à la sortie du docking, soit après des étapes post-docking telles que : minimisation, minimisation des résidus, minimisation du backbone de la protéine, optimisations des liaisons hydrogène.
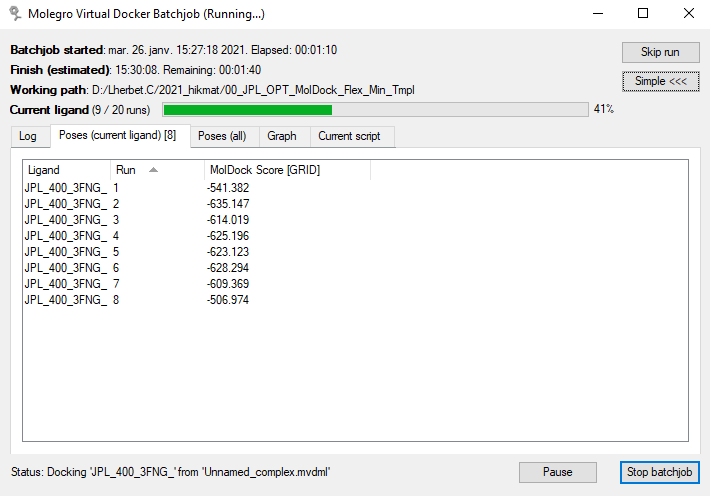 |
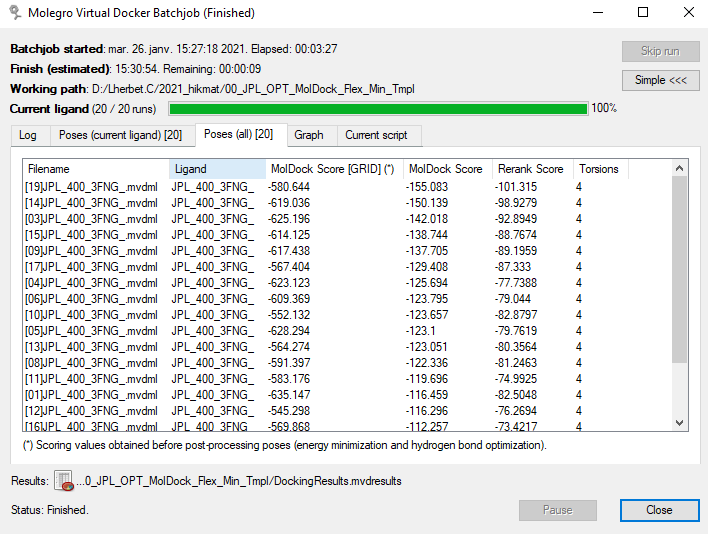 |
| Fin du docking, classement par défaut : ordre de calcul (numéro de Run). | Après des étapes post-docking internes, classement par valeur de MolDock Score. |
Si nous prenons la même pose : le Run 2 (fin du docking) et la pose [01] (post-docking) nous avons une valeur de MolDock Score [GRID] (‘MolDockGrid’ dans les paramètres) égale à -635.147 (valeur arbitraire). Rappelons qu’il s’agit du score utilisé pour le docking, approximé sur une grille de 0.3 Å (valeur réglable) pour accélérer les calculs. La valeur ‘réelle’ explicite du score MolDock après les étapes post-docking est de -116.459 pour cette pose. Le score Rerank est un autre système de score, basé sur un re-paramétrage pondéré de MolDock, et qui est systématiquement calculé.
3. Suivi de la convergence
Cette manipulation in silico a été paramétrée pour 20 runs, en fonction des paramètres de clustering, et les deux se sont avérés suffisants pour échantillonner l’espace conformationnel. Ce n’est pas toujours le cas, parfois il faut augmenter significativement le nombre de runs (donc de poses, donc le temps de calcul).
Il existe une relation entre le nombre de runs et la valeur limite (minimale) du score que l’on peut atteindre. La connaissance (tracé) de cette fonction permet de savoir, d’une manière objective, si le nombre de runs est acceptable (pour un ligand donné). On cherchera toujours (ou quand on le peut) à atteindre une valeur du score limite. Certains logiciels nous donnent les moyens de faire cette recherche (c’est le cas de Molegro) d’autres non.
Dans ce contexte, l’onglet Graph est très intéressant, il permet de visualiser l’optimisation en cours. L’image suivante montre son contenu pour un calcul réalisé avec un autre algorithme, de type OPT (dans le cas MSE le graphe sera différent) :
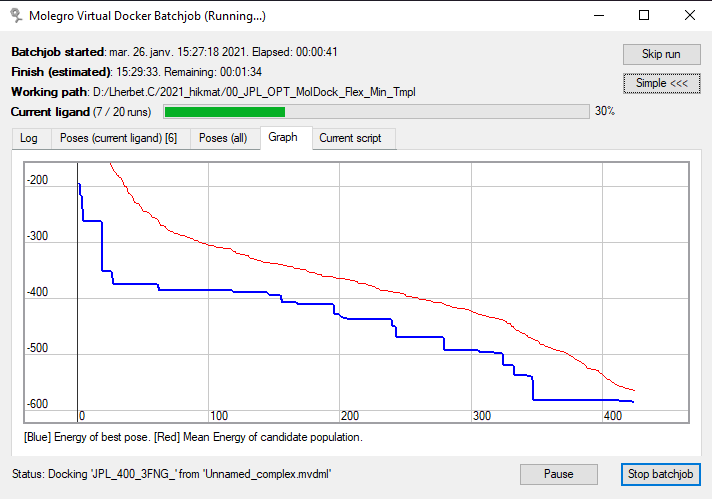 |
Il s’agit de la valeur du score [GRID] en fonction du nombre de pas de calculs. Son apparence dépendra des spécificités de l’algorithme. Dans ce cas, lorsque la courbe rouge rejoint la courbe bleue, et que les deux restent ensemble pendant les itérations suivantes, la convergence est atteinte. Il n’est plus possible d’améliorer les résultats (la meilleure valeur du score possible est atteinte) sur ce run, le calcul s’arrête et le programme passe au ligand suivant.
Dans les paramètres le nombre d’itérations maximum (maxIterations=10000) est fixé. Dans ce cas, la convergence était atteinte avant 1000 pas de calculs, il y a donc de la marge. Sauf que le JPL est une molécule ‘facile’ avec des cycles aromatiques lignés et 4 angles de torsion. D’autres molécules peuvent être plus complexes, notamment si le nombre d’angles de torsion augmente (ex: branchement d’espaceurs ou de chaînes carbonées). Ces molécules nécessitent plus de pas de calcul ou une population plus grande (il s’agit d’un algorithme évolutionnaire).
La convergence est importante pour nos calculs, même si cet aspect est tempéré par des calculs post-minimisation (optimisation du ligand et des interactions, minimisation des chaines latérales et du backbone). Elle l’est moins pour du criblage, ou c’est un signal qui est recherché avant une conformation optimale ou une pose forte (nous reviendrons sur ce concept dans un autre article). Nous chercherons toujours (ou quand on le peut) à l’atteindre avant la valeur de maxIterations. Certains logiciels nous donnent les moyens de faire ce type de vérification, mais beaucoup d’autres ne le font pas.
4. Suivi des calculs
Une manière de faire le suivi des calculs est de regarder la charge du processeur, en voici un exemple (en mode CPU exclusif, pas de calculs GPU) lors du lancement du script :
 |
Un calcul Molegro plus les processus Windows occupent 30% du processeur. Il s’agit d’un CPU (type Intel Xeon d’ancienne génération) avec 4 cœurs. Sur ce type de processeur on peut encore lancer 3 calculs, chaque calcul (un lancement de script) prenant environ 20% du CPU. Chaque calcul étant indépendant, la relation est presque linéaire (au moins jusqu’à 7-8 calculs en simultanés) en fonction du nombre de cœurs et de sockets, de la mémoire (8 à 16 Go suffisent), et de l’architecture (bus, organisation mémorielle, disques, etc).
Il existe un fonctionnement plus avancé (grille) de Molegro en termes de parallélisme, mais ce n’est pas l’objet de l’article. Dans le cadre de calculs au quotidien, avec des paramètres bien optimisés, l’étape limitante en temps n’est pas le calcul (car on calcule mieux) mais l’analyse et l’interprétation des résultats.
5. Conclusion
Avec Molegro nous constatons que le paramétrage est riche (plus de 25 valeurs) et accessible. Ce qui permet de se sortir de situations impossibles auxquelles sont confrontés d’autres programmes en ‘boite noire’ (ça passe ou ça ne passe pas). Des fichiers standards, PDB, SDF, sont utilisés, il n’est pas besoin de générer des fichiers intermédiaires, tels que PDBQT (PDB : Protein Data Bank, Q :Partial Charge, T : Atom Type) comme avec certains outils.
La combinaison entre { nombre de runs, convergence, algorithme et fonction de score } est assimilable (pour donner une image) à la résolution d’un instrument optique. En connaissance de ces limites et de celles imposée par la facilité d’utilisation, il faudra donc savoir ce que l’on peut faire avec l’outil et adapter le champ d’études en fonction de la ‘résolution’ disponible.